
Mise à jour Doc, correction du jnlp pour inclure Mac OS X
git-svn-id: https://subversion.renater.fr/xtandempipeline/trunk@99 b8ef2a07-7df7-436f-90b9-41648038564b
Showing
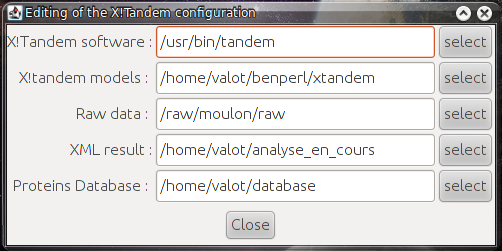
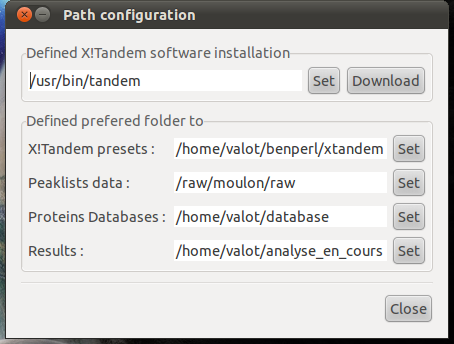
- xtandempipeline/doc/images/tandem_configuration.png 0 additions, 0 deletionsxtandempipeline/doc/images/tandem_configuration.png
- xtandempipeline/doc/images/tandem_export.png 0 additions, 0 deletionsxtandempipeline/doc/images/tandem_export.png
- xtandempipeline/doc/images/tandem_filter.png 0 additions, 0 deletionsxtandempipeline/doc/images/tandem_filter.png
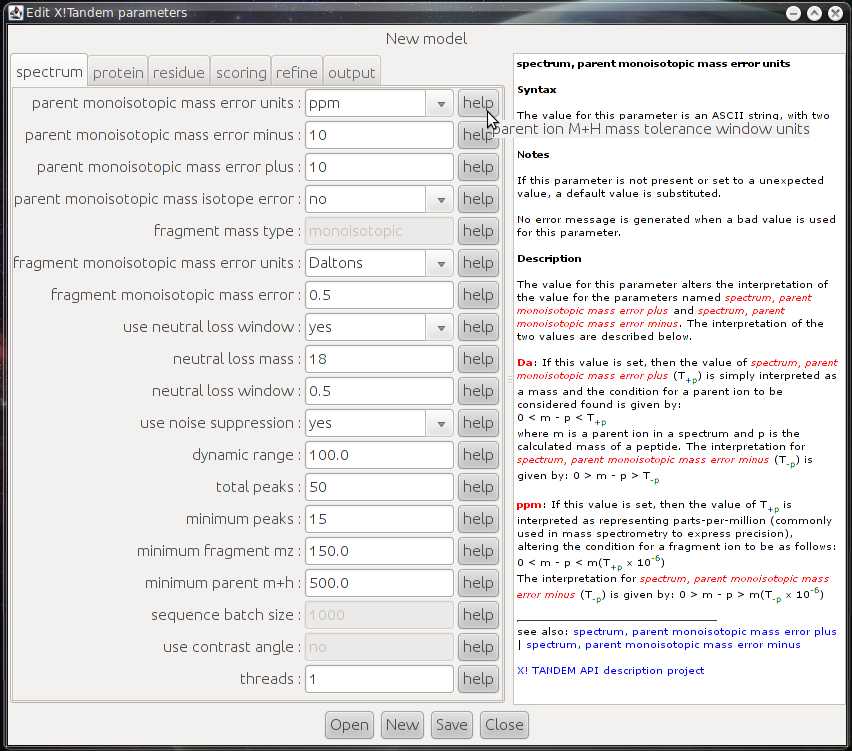
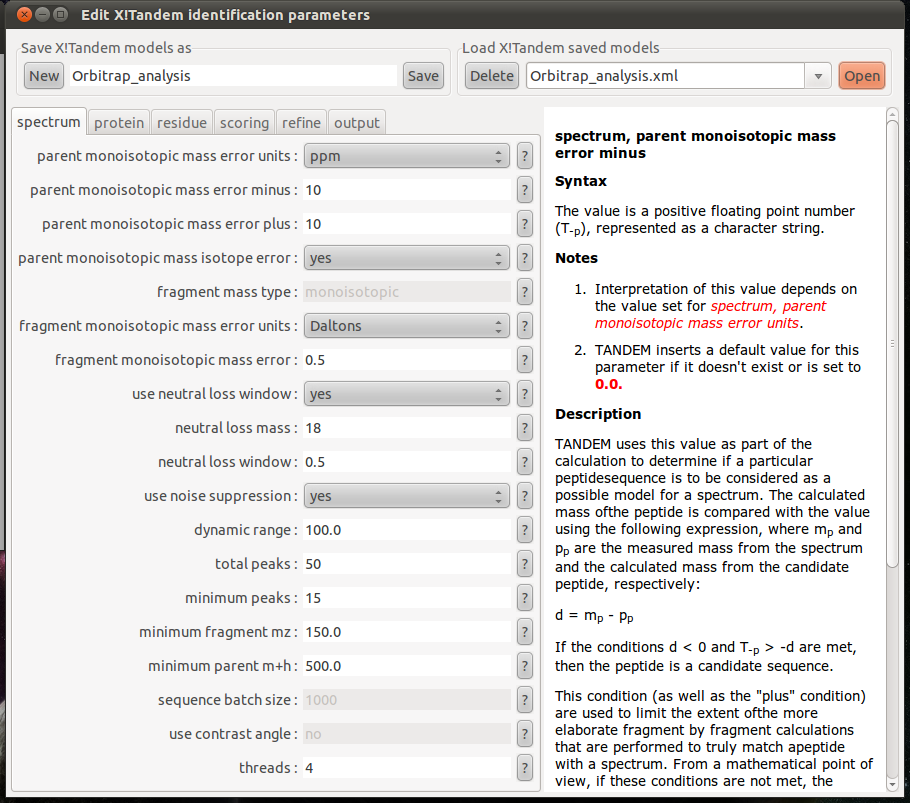
- xtandempipeline/doc/images/tandem_parameter.png 0 additions, 0 deletionsxtandempipeline/doc/images/tandem_parameter.png
- xtandempipeline/doc/images/tandem_principal.png 0 additions, 0 deletionsxtandempipeline/doc/images/tandem_principal.png
- xtandempipeline/doc/xtandem_pipeline.pdf 0 additions, 0 deletionsxtandempipeline/doc/xtandem_pipeline.pdf
- xtandempipeline/doc/xtandem_pipeline.tex 109 additions, 50 deletionsxtandempipeline/doc/xtandem_pipeline.tex
- xtandempipeline/lib/swt/swt-cocoa-macosx-x86.jar 0 additions, 0 deletionsxtandempipeline/lib/swt/swt-cocoa-macosx-x86.jar
- xtandempipeline/lib/swt/swt-cocoa-macosx-x86_64.jar 0 additions, 0 deletionsxtandempipeline/lib/swt/swt-cocoa-macosx-x86_64.jar
- xtandempipeline/share/xtandempipeline/xtandempipeline.jnlp 15 additions, 7 deletionsxtandempipeline/share/xtandempipeline/xtandempipeline.jnlp
{kind=link}
{kind=link}
| W: | H:
| W: | H:
{kind=link}
{kind=link}

| W: | H:

| W: | H:
{kind=link}
{kind=link}

| W: | H:

| W: | H:
{kind=link}
{kind=link}
| W: | H:
| W: | H:
{kind=link}
{kind=link}
| W: | H:
| W: | H:
No preview for this file type
File added
File added